
Le Neoplasie Endocrine Multiple (MEN)
Le Neoplasie Endocrine Multiple (MEN) sono delle sindromi rare caratterizzate dalla presenza di iperplasie/neoplasie di numerose ghiandole endocrine e si distinguono in MEN di tipo 1, MEN di tipo 2, MEN di tipo 2B (MEN3) e MEN 4 . Tali sindromi si presentano frequentemente in forma familiare a trasmissione autosomica dominante. In questo caso il rischio per un genitore affetto di trasmettere la patologia alla progenie è del 50%.
Le recenti acquisizioni nel settore della genetica molecolare hanno reso possibile per ambedue le patologie l’esecuzione di test genetici per l’individuazione del/i portatore/i del gene mutato. Sono altresì stati messi a punto protocolli diagnostici, sia strumentali che boichimici, che permettono un precoce riconoscimento delle patologie endocrine tipiche delle sindromi MEN. Inoltre la terapia medica e chirurgica per le singole endocrinopatie rende oggi possibile al paziente una sempre migliore qualità di vita.
Numerose sono le specialità mediche interessate a tali patologie ed il paziente si troverà a rivolgersi inizialmente non solo all’endocrinologo ma anche ad altri specialisti. E’ peraltro importante che il sospetto diagnostico nasca nel medico di famiglia che potrà così indirizzare il paziente verso un iter diagnostico rapido ed efficace.
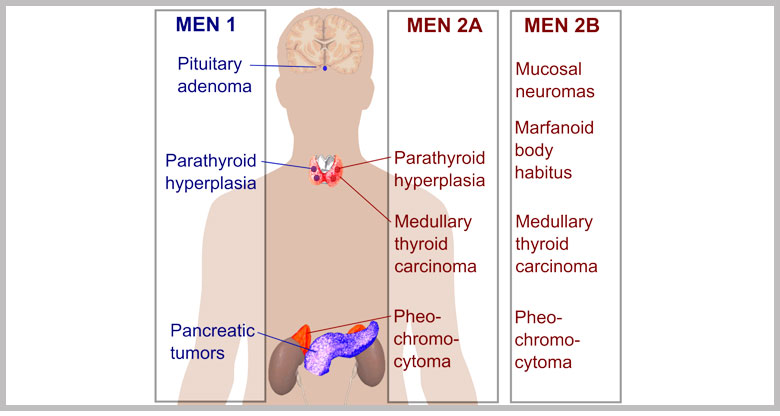
MEN 1
E’ anche indicata come adenomatosi endocrina multipla familiare o sindrome di Wermer. E’ una malattia ereditaria, trasmessa con meccanismo autosomico dominante. E’ un disordine piuttosto raro, con una prevalenza di circa 3-20/100.000 individui. Colpisce ambedue i sessi equamente e non dimostra alcuna preferenza geografica, razziale od etnica. E’ causata da una combinazione di più di 20 tumori endocrini e neuroendocrini ed è pertanto complicato darne una definizione precisa. Praticamente la MEN1 viene definita come la presenza di un caso con 2 dei 3 principali tumori endocrini MEN1-relati (adenomi paratiroidei, tumori neuroendocrini entero-pancreatici ed adenomi ipofisari). Si parla di MEN1 familiare invece, in presenza di almeno 1 caso di MEN1 più almeno 1 parente di primo grado con 1 di queste 3 tumori.
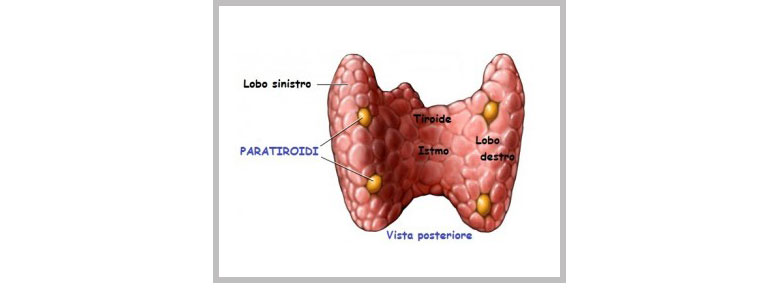
Le PARATIROIDI sono le ghiandole più frequentemente interessate in questa sindrome. Le ghiandole paratiroidee sono comunemente 4 e sono localizzate in prossimità della ghiandola tiroide. Le paratiroidi producono un ormone chiamato paratormone (PHT), che controlla la concentrazione di calcio nell’osso, nel sangue e nelle urine.
Nella MEN 1 le 4 ghiandole paratiroidee tendono ad essere iperattive, con conseguente iperparatiroidismo, caratterizzato da un eccesso di PTH che causa l’aumento del calcio nel sangue (ipercalcemia).
L’iperparatiroidismo primitivo è il disturbo più comune nella MEN 1, raggiungendo quasi il 100% di penetranza all’età di 50 anni. Al contrario la MEN 1 è una patologia estremamente rara, rappresentando solo il 2-4% di tutti i casi di iperparatiroidismo primitivo. Tale condizione generalmete è la prima espressione clinica della MEN 1, con una tipica età di insorgenza intorno ai 20-25 anni, e può rimanere silente per molti anni prima di essere diagnosticata, anche casualmente, nel contesto di uno screening biochimico. L’ipercalcemia non riconosciuta può determinare un carico eccessivo di calcio a livello renale con un incremento dell’escrezione urinaria di tale ione e conseguente formazione di calcoli renali o di nefrocalcinosi.
All’iperparatiroidismo si accompagnano sintomi quali facile affaticabilità, astenia, dolori muscolari o ossei, stitichezza, difficoltà digestive, calcoli renali e/o diminuzione della massa ossea.
Talvolta è difficile decidere se l’iperparatiroidismo associato alla MEN 1 necessiti di un trattamento immediato, specialmente in un soggetto che non mostra sintomi. Il trattamento di scelta consiste in un intervento chirurgico che rimuova le 4 ghiandole paratiroidee; in alcuni centri la paratiroidectomia totale è poi seguita dal successivo reimpianto di frammenti di tessuto paratiroideo nell’avambraccio, per evitare che il paziente diventi ipotiroideo e per rendere l’eventuale asportazione in caso di recidiva di iperparatiroidismo più semplice ed immediata.
Qualora, si ricorresse alla paratiroidectomia totale, sarà indispensabile assumere quotidianamente una terapia sostitutiva a base di calcio e di vitamina D per prevenire la diminuzione del calcio nel sangue (ipocalcemia). Esiste inoltre, un farmaco calciomimetico (cinacalcet) da utilizzare qualora non fosse indicato l’intervento chirurgico di paratiroidectomia in caso di gravi comorbidità.
Altro organo frequentemente interessato nella MEN 1 è il PANCREAS che oltre a rilasciare succhi digestivi nel tubo digerente, secerne anche sostanze ormonali nel torrente circolatorio (pancreas endocrino). Il tumore entero-pancreatico più frequentemente riscontrato nella MEN 1 è il gastrinoma (sindrome di Zollinger-Ellison), con successiva ipersecrezione di gastrina, un ormone che circola normalmente nel sangue, determinando una produzione acida gastrica sufficiente per la digestione. Se esposto ad un eccesso di gastrina, lo stomaco rilascia, a sua volta, un eccesso di acido cloridrico che porta alla formazione di ulcerazioni gastroduodenali multiple oltre a causare diarrea. Peraltro, i gastrinomi in MEN 1 hanno preferenzialmente sede duodenale dove si sviluppano a livello della sottomucosa. Le ulcere causate dai gastrinomi sono più pericolose delle tipiche ulcere gastriche o duodenali essendo multiple e recidivanti. Se non trattate possono causare perforazioni dello stomaco e/o dell’intestino con conseguenze molto gravi. Circa 2/5 dei gastrinomi sono presenti nei pazienti affetti da MEN 1. Alla diagnosi circa la metà sono metastatici già alla diagnosi. La terapia dei gastrinomi è farmacologica con gli inibitori di pompa protonica (PPI), bloccanti la produzione acida gastrica, ma anche chirurgica.Avendo questi tumori nella MEN 1 un’origine frequentemente duodenale, attualmente l’intervento chirurgico consiste nell’asportazione del duodeno e della testa pancreatica (duodeno-cefalopancreasectomia). Per tutti gli altri tumori pancreatici secernenti, il trattamento di scelta è sempre quello chirurgico.
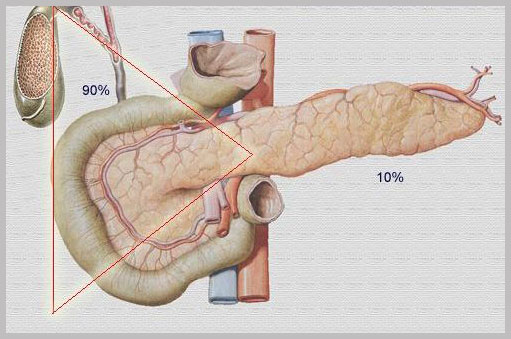 Trinagolo del gastrinoma
Trinagolo del gastrinoma
.
Altri tipi di tumori neuroendocrini pancraetici associati a MEN 1, seppur meno frequenti dei gastrinomi, sono gli insulinomi, i glucagonomi, i somatostatinomi, i VIPomi ed i pNET che secernono PP (polipeptide pancreatico). I tumori pancreatici endocrini nella MEN 1 possono anche non produrre ormoni, parleremo in questo caso di pNET non secernenti.
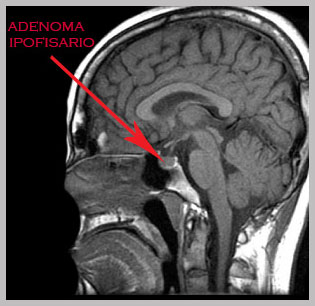
Gli adenomi ipofisari sono la prima manifestazione clinica della MEN 1 nel 25 % dei casi. La loro prevalenza nella MEN 1 va dal 10 % al 60 %. Circa 2/3 sono microadenomi (<1 cm), Per quanto riguarda gli adenomi ipofisari associati a MEN 1 i più frequanti sono i PRLomi, ma si possono osservare anche adenomi ipofisari GH-secernenti, ACTH-secernenti, TSH-secernenti, LH/FSH-secernenti.
Neoplasie più rare associate alla MEN 1
Nella MEN 1 oltre alle neoplasie sopracitate più frequentemente riscontrate, si possono diagnosticare anche altri tumori, quali lipomi cutanei (30%), carcinoidi timici, bronchiali ed intestinali (2%), ependimomi (1%), feocromocitoma (<1%), angiofibromi del volto (85%), collagenomi (70%), adenomi surrenalici.
Diagnosi genetica
Il gene MEN 1 è localizzato sul cromosoma 11 e codifica per una proteina ad azione oncosoppressiva, detta menina di 610 aminoacidi espressa in molti tessuti, a localizzazione nucleare nelle cellule quiescenti e citoplasmatica nelle cellule in divisione. La Menina interagisce con il complesso trascrizionale AP1, in particolare con il fattore JunD, regolando in modo negativo la progressione del ciclo cellulare. JunD è un soppressore della crescita cellulare; In condizioni patologiche, lega la proteina menina mutata e si verifica uno switch di funzione assumendo le funzioni di promotore della crescita cellulare. Sono state riportate più di 1300 mutazioni in letteratura. Si tratta di diversi tipi di mutazioni, approssimativamente nel 25% dei casi si tratta di mutazioni nonsense, nel 45% dei casi di piccole delezioni, nel 15% di piccole inserzioni, in meno del 5% di mutazioni donor-splice e nel 10% di mutazioni missense. Tutte queste mutazioni possono generare una proteina (menina) assente o troncata, provocando una perdita della funzione oncosoppressiva della menina, con conseguente sviluppo del tumore.
MEN 2
E’ una rara malattia ereditaria, caratterizzata dallo sviluppo di tre specifiche malattie endocrine: il carcinoma midollare della tiroide (MTC), il feocromocitoma e l’iperplasia delle paratiroidi. Il rischio di sviluppare una o più manifestazioni cliniche, nonché l’età di insorgenza dipende dalla mutazione presente nel gene responsabile della malattia chiamato proto-oncogene RET; la sindrome è caratterizzata da un’alta correlazione genotipo-fenotipo, ovvero specifiche alterazioni genetiche determinano quadri clinici caratteristici e prevedibili. Dal punto di vista clinico si distinguono tre varianti della sindrome denominate MEN2A, MEN2B (MEN 3) e FMTC.
MEN 2A
Si tratta di una patologia autosomica dominante, ad insorgenza nell’età giovane adulta, nel 95% dei casi sono presenti carcinomi midollari della tiroide (MTC) spesso multifocali, accompagnati sempre da iperplasia delle cellule C, nel 50% dei casi feocromocitoma (pheo) (surrenalico monolaterale o bilaterale), nel 15% dei casi iperparatiroidismo (HPT) (molto più frequente l’iperplasia rispetto all’adenoma); raramente il quadro può essere accompagnato da lichen amiloidosico interscapolare, malattia di Hirschprung, nervi corneali prominenti:
- MEN 2A-1: MTC + pheo + HPT
- MEN 2A-2: MTC + pheo
- MEN 2A-3. MTC + HPT
MEN 2B (MEN 3)
È una patologia ad insorgenza precoce del MTC nel 90% dei casi (spesso multifocale e associato a iperplasia delle cellule C) associata o meno a feocromocitoma (nel 50% dei casi) + dismorfismi facciali, neurinomi delle mucose (labbra e lingua), habitus marfanoide, cifoscoliosi, ganglioneuromatosi intestinale, pectus excavatum, pes cavus.
FMTC
Carcinoma midollare della tiroide familiare: almeno 4-8 casi di MTC in famiglia ad insorgenza in età giovanile o adulta, non feocromocitoma né HPT.
Diagnosi genetica
Il gene RET (10q11.2) codifica per un recettore di membrana ad attività Tyrosin Kinasica (114 aminoacidi) presente sulla membrana di alcune cellule derivanti dalle creste neurali (cellule parafollicolari C, cellule gangliari della midollare del surrene, gangli gastroenterici). Il legame con il recettore comporta la dimerizzazione di due recettori con reciproca fosforilazione determinando un segnale proliferativo. Nel caso della MEN2 si verificano mutazioni missense che determinano attivazione del recettore RET. Le mutazioni più comuni della proteina RET correlate alle manifestazioni tumorali sono:
- MEN2A: sostituzione di una cisteina in posizione 634
- MEN2B: sostituzione in posizione 918 di una metionina con una treonina
- FMTC: sostituzioni cisteine in altre posizioni e sostituzioni ai domini TK
MEN 4
Recentemente studi effettuati hanno mostrato una variante della sindrome MEN insorta spontaneamente in una colonia di ratti. Gli animali affetti sviluppavano tumori endocrini multipli, con uno spettro di caratteristiche simili sia alla MEN1 che alla MEN2 umane (adenomi ipofisari multifocali, feocromocitoma bilaterale, paragangliomi, iperplasia cellule C, adenomi a carico di paratiroidi e NET pancreatici). Sono state cercate e trovate, sebbene in un numero esiguo di casi, mutazioni della p27 in pazienti con caratteristiche fenotipiche della MEN1 ma negativi per la mutazione. Proteina P27 è un inibitore della ciclina chinasi-dipendente, coinvolta nei processi di regolazione del ciclo cellulare e della proliferazione cellulare. Quindi è stata identificata una nuova sindrome MEN nell’uomo, denominata MEN4, causata da mutazioni nel gene CDKN1b/P27, che deve pertanto essere considerato un nuovo gene di suscettibilità per lo sviluppo di tumori endocrini multipli. Per la MEN 4, visto l’esiguo numero di casi ad oggi descritti, non è possibile attualmente definire un quadro clinico specifico, né sono ancora presenti linee guida per lo screening biochimico e strumentale, pertanto vengono seguiti come pz MEN1 e MEN2 like.